
Pharmacovigilance (PV) is defined by the World Health Organization (WHO) as the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug-related problem. PV focuses on adverse drug reactions, or ADRs, which are defined as any response to a drug which is noxious and unintended, including lack of efficacy.
Ultimately, PV is concerned with identifying the hazards associated with pharmaceutical products and with minimizing the risk of any harm that may come to patients. Sounds simple enough, but the process that pharmaceutical companies go through to satisfy this requirement is rather complex. But first, a brief history of Pharmacovigilance. I’ve included a helpful timeline below.
Historic Timeline of Pharmacovigilance
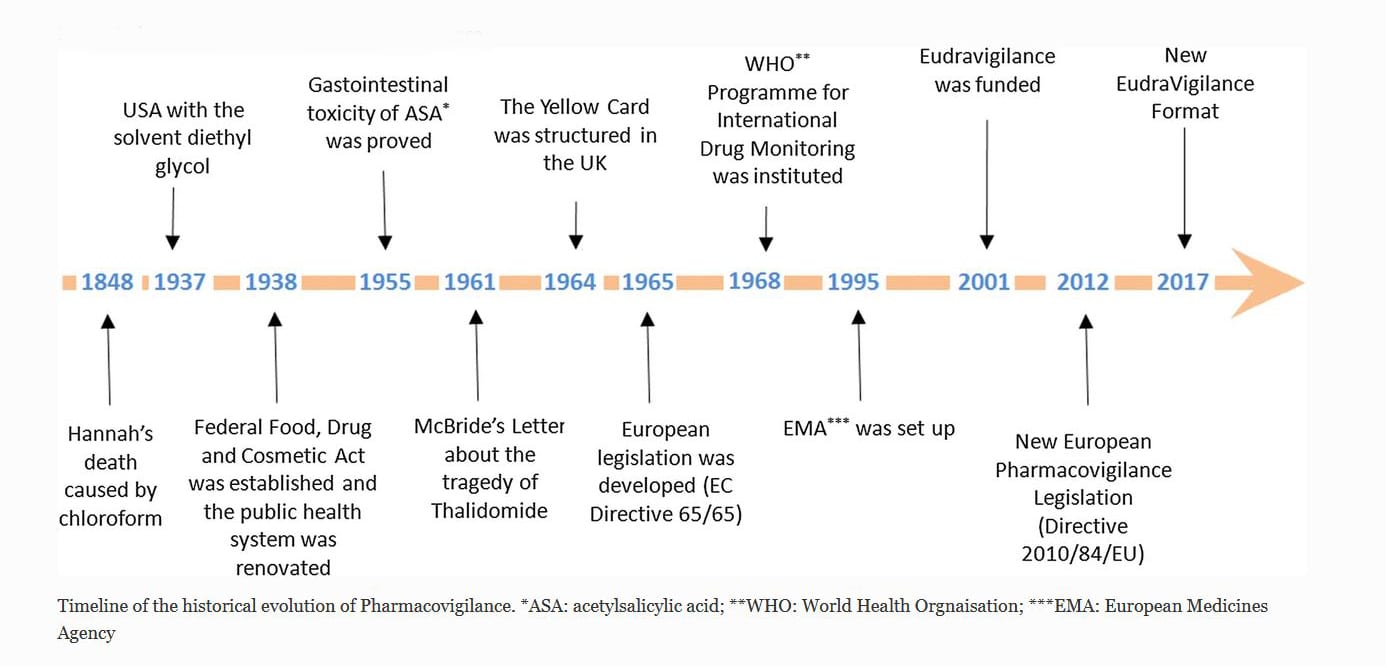
While there are many significant historical events that contributed to the genesis of PV, these milestones shown above are just a few of the significant ones. One of the earlier reported events was when a young girl, Hannah, died after receiving chloroform anesthetic before the removal of an infected toenail. While the cause of death was not determined at that time, it did begin to raise awareness of adverse events related to anesthesia. It’s interesting the think about how many centuries went by without this type of detection or oversight.
An Overview of Important Legislation and Regulations in Pharmaceutical History
US and FDA Origins
The US Federal Food and Drug Act was formed on June 30, 1906 with the passage of the 1906 Pure Food and Drugs Act. This law prohibited interstate commerce in adulterated and misbranded food and drugs, and provided the basic elements for consumer protection. Additionally, in 1911, the organization began to regulate against the false therapeutic drug indications.
In 1937, there were more than 100 deaths associated with a solvent used in a sulfanilamide elixir. Public outcry lead to the creation of the Food, Drug and Cosmetic Act. Its aim was to renovate the public health system and introduce protections regarding the safety of drugs before their market approval.
WHO and EMA Origins
European Pharmacovigilance changed significantly due to what is known as the tragedy of Thalidomide, or the Thalidomide Disaster, in 1961. Dr. McBride, an Australian doctor, observed a connection between congenital malformation of babies and thalidomide. In his letter to the editor of the Lancet Journal, he outlined the incidence of congenital malformations of babies; 1.5% had increased up to 20% in women who had taken thalidomide, that was marketed as a sleeping pill, during pregnancy [1]. The tragedy of thalidomide raised issues around the reliability of tests, company operations, and the importance of monitoring the drugs after their release to the market.
In 1968, the WHO Program for International Drug Monitoring was formed, and in 1995, the European Medicines Agency (EMA) was created. While there were other important legislative and regulatory updates, one of the key updates was the formal definition for an Adverse Event as “a response to a medicinal product which is noxious and unintended.” This encompassing definition covers any adverse event following the use of a medicine including medication errors and uses outside the terms of the authorized use.
All these events and more paved the way for increased awareness regarding the formality of Adverse Event Reporting and the marketing companies’ responsibilities regarding drug safety. The importance of this evolution cannot be stressed enough. The clients we partner with in the mission of Life Sciences are directly impacted by this practice too, which is executed through a complex process.
Sources:
[1] McBride WG. Thalidomide and congenital abnormalities. Lancet. 1961; ii:1358.

